loading...
分子动力学优化方案
背景简介
分子动力学模拟技术是研究分子之间运动的一种工具,是典型的计算瓶颈问题。组成物质的微观粒子的数量十分庞大,且为精确跟踪粒子的运动,保证数值模拟的稳定性,必须使用很小的时间步长,计算量十分庞大。所以分子动力学模拟对计算机性能的需求是无止境的,并行计算是必由之路。
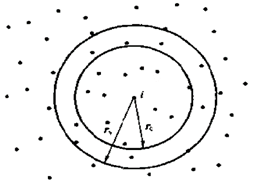
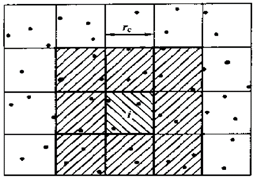
通常情况下会采用设定邻近列表(图1)和将计算空间分解成若干个元胞的优化方法(图2)。在模拟过程中,受力计算是最费时的,占了整个模拟的90%左右,是优化实现的重点。
优化介绍
为了加速计算受力时寻找原子对的过程,分子动力学模拟经常采用邻近列表方法,对每个原子建立一个邻近列表,然后将符合条件的原子加入邻近列表中。计算力的算法常采用牛顿第三定律。
经典的并行化方法有域分解、任务分解、流水线等,在分子动力学模拟上采用流水线方法会有更好的表现,将计算机指令处理过程拆分为多个步骤,通过多个处理单元并行执行加快指令执行速度。SIMD指令集的使用,把64位寄存器拆成8个8位寄存器同时完成8个操作,理论计算效率会提升8倍。
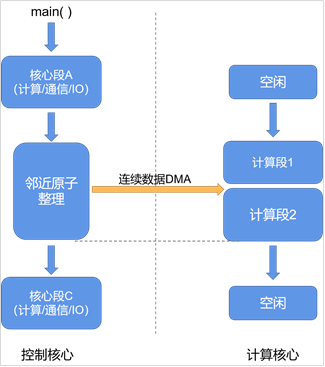
针对某异构众核处理器平台,计算核心的超强浮点计算能力。通过异步并行方法(图3),达到计算隐藏效果,提高运行效率;解决邻近列表中原子不连续,离散访存的问题,减少1/4的数据量,增加数据复用。
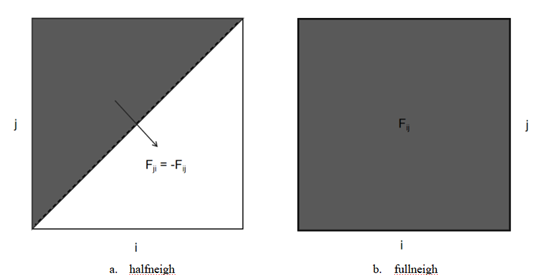
改用完整的邻近列表(图4)增加计算,降低数据关联性,减少频繁的查表操作;引入原子簇的概念,相邻的所有粒子组成一个簇,共享大部分邻近列表,减少将近20%的邻近原子,使邻近列表的建立时间缩减30%。
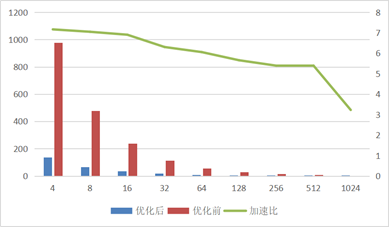
达到整体加速将近8倍(图5),解决合作用户的课题研究。不断深入研究与优化,突破超大并行规模瓶颈,在1024x1024x1024的原子规模下,实现了整体10倍以上的加速效果,且并行规模可扩展到千万核心(图6)。为分子模拟、材料领域内的计算模拟提供相关软件,缩短模拟研究周期。